Let’s take an excursion outside of “Understanding and Using the Statistics of Communication Signals” by looking at a naturally occurring signal: the human genome.
Anyone can download a complete sequence of nucleic acids that make up the twenty-three human chromosomes at a website maintained by the National Center for Biotechnology Information, which is a branch of the National Institutes for Health in the USA.
Adding up the number of nucleic-acid pairs across the chromosomes yields a sequence of about three billion letters–A, C, G, T–for humans, and other organisms have more or less.
The four letters correspond to four nucleic acids: adenine (A), cytosine (C), guanine (G), and thymine (T). So the genome is a sequence of these acids, arranged in pairs in a double helix, and broken up into separate physical chunks called chromosomes.

But me, I see a signal. And so I want to process that signal with my CSP tools to see what might be revealed that is not obvious from simpler processing, such as spectrum estimation or autocorrelation estimation.
The Downloaded Files
I’ve downloaded two files: GRCh37_latest_genomic.fna and GRCh38_latest_genomic.fna. These are giant text files that consist of the four ASCII characters G, T, A, C, but also g, t, a, and c. There are some stretches of the files that contain sequences of the letter N as well. Why the upper and lower case versions of the four nucleic acids? It isn’t completely clear to me, but it seems to have to do with repeated sequences throughout the genome. Try to figure it out here. Explanations are welcome in the Comments below.
The N character is used when the acids are not clearly identified by whoever is doing the identification. They are fairly rare and I just skip over them and continue encoding the letters into numbers (see below) without introducing any intervening numbers. A typical stretch of letters from the downloaded files is shown in Figure 2.

Converting from DNA Letters to Numbers
Now none of my signal-processing tools can process a sequence of letters, so we have to choose a mapping between the four nucleic acids G, A, T, and C and real or complex numbers. The problem is a bit more involved than that, though, because the DNA files contain the four letters G, A, T, and C but also g, a, t, and c, their lowercase versions, as we saw above.
I first tried to encode the letters using a QPSK constellation, and I treated the uppercase and lowercase letters as identical. That produced some mildly interesting spectral correlation plots, but later I tried an 8ASK constellation, where I treat the uppercase and lowercase separately. Maybe that isn’t cool–let me and my readers know if you think that was a mistake.
The 8ASK encoding is: t = -7, T = 7, a = -5, A = 5, c = -3, C = 3, g = -1, and G = 1. I haven’t encoded the entire genome file, just the first 
CSP Video
Once I had the nucleic acids coded as integers, I had a signal I could process! As usual at the CSP Blog, I used a sliding-block style of processing and assembled the results for each block into a movie. The blocks are 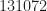

We are not processing a function of time here, but of space. Figure 1 comes in handy. Normally we use a sampling rate that is expressed in units of Hertz, which arises from a sampling increment–the separation in time between two successive time-series (signal) values–that is expressed in units of seconds. Here for the genome we have a ‘sampled’ signal that corresponds to a sampling rate expressed in units of reciprocal meters (rm), which arises from a sampling increment–the separation in space between two successive distance-series (signal) values–that is expressed in units of meters. So our sampling increment is 

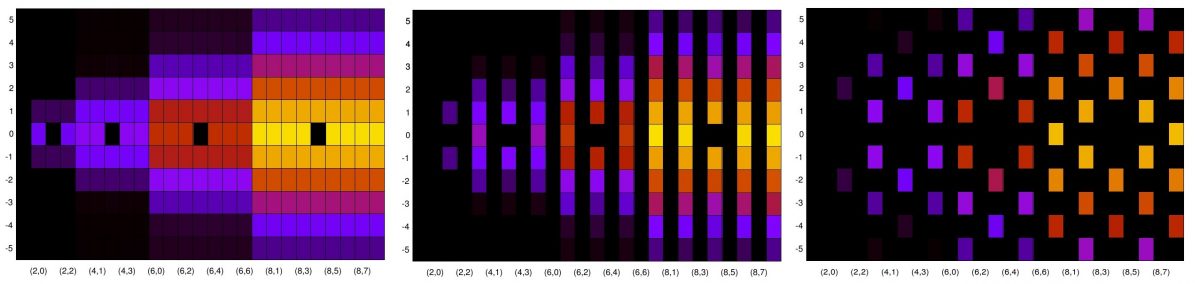
The blind CSP applied to GRCh37_latest_genomic.fna is shown in Video 1. For each block, I first apply a sine-wave detector to determine if there are any additive sine-wave components. If so, I remove them prior to CSP. The sine-wave-expurgated block is then used as input to the SSCA, producing estimates of the cycle frequencies for the block. These are then used by the FSM to estimate the spectral correlation function, and these spectral correlation function slices are arranged and shown in the uppermost plot. I’ve reduced the magnitude scale to focus on the non-zero cycle frequencies at the expense of truncating the PSD.
There are many fascinating frames in the movie, and many cycle frequencies are detected. What do they mean? I don’t know.
A particularly intriguing frame occurs around 

Anyway. This is desultory, so that’s all I have for now.
Comments appreciated!
Now this is an interesting post. One of the hard parts about describing CSP is presenting a “real world” example that is both cyclostationary and easy to understand; that usually ends up being the temperature. But this is a fascinating example I had not even considered. Thank you!